How it works? Get started with CYCLOPEp
1. Choose between the available types of CP, combining alpha, beta, gamma aminoacids. Currently, only working for alpha-D,L-CPs
2. Number of CPs: Begin by selecting the total number of CP sequences you need for your design from a dropdown number selector. 3. CP Sequences: Input your CP sequences using the standard amino acid letter code, ensuring they consist of an even number of amino acids, between 4 to 12. The sequence by default starts by D (alternating with L, D, L‚etc..) If you're designing more than one CP, additional input fields will appear accordingly. Alongside each new sequence field, specify the relative orientation-parallel or antiparallel-to the preceding sequence. 4. Force Field Selection: Choose the appropriate force field for your design from a dropdown menu, which will be used for parametrizing your CPs. 5. Name Your Design: Assign a unique name to your project or let the system generate one based on the force field and sequence combination.
Outcome?
Upon submission, CYCLOPEp Builder will provide you with the PDB files for your designed CPs and the nanotube. Additionally, you will receive the ITP files configured for the selected force field, ready for use in GROMACS.Begin your peptide design journey with CYCLOPEp Builder today!
A bit of history
Ghadiri’s group pioneered the synthesis of Cyclic Peptides in 1993, utilizing a balanced alternation of D and L-α residues. This unique chirality forms a flat ring structure, positioning the amino and carbonyl groups of the peptide backbone perpendicular to the ring's plane. Such a structure promotes the self-assembly of CPs into SCPNs through hydrogen bonding, adopting either a parallel or antiparallel β-sheet-like orientation. Intriguingly, the amino acid side chains protrude outward from the Nanotube, influencing its properties. SCPNs crafted from hydrophobic CPs tend to form transmembrane nanotubes. In contrast, amphipathic variants reside atop membranes, showcasing potent antimicrobial actions.
Enhancing Cyclic Peptide Research through Molecular Dynamics Simulations
Research on cyclic peptides (CPs) and self-assembled cyclic peptide nanotubes (SCPNs) often faces significant challenges
in achieving precise structural characterization. Traditionally, methods like NMR spectroscopy are used, but these
techniques frequently struggle to capture the various conformations that CPs and SCPNs can assume in solution, which
complicates a full understanding of their properties. Molecular dynamics (MD) simulations emerge as a potent complement
to these traditional methods, offering a dynamic and detailed view of molecular behaviors that are otherwise
inaccessible.
CYCLOPEp supports this research process by providing researchers with the necessary files to conduct their own MD
simulations. Equipped with these MD simulation-ready files, researchers can accurately predict crucial experimental
observables such as thermodynamic stability, reaction kinetics, and binding affinities. This not only deepens the
theoretical understanding of CPs and SCPNs but also improves experimental planning. By enabling targeted experimentation
on the most promising molecular conformations, CYCLOPEp helps optimize resource use and accelerates discoveries in
cyclic peptide-based research.
All-Atoms, is Precision everything?
Cyclic Peptides (CPs) are primarily characterized by the directionality of their hydrogen bonds, which facilitates their parallel/antiparallel self-assembly. In all-atom simulations, every atom corresponds to a single particle in the force field, offering a detailed representation of molecular interactions. Yet, the granularity comes at a cost; with a time step as brief as 2fs, these simulations become computationally demanding, especially for processes like self-assembly that unfold over microseconds. Given these challenges, Coarse-Grain simulations emerge as a compelling alternative.
Martini and the Chirality problem
MARTINI stands out as a prevalent Coarse-Grain ForceField used in MD simulations of biomolecular systems. Its model adopts a four-to-one mapping approach, meaning that, on average, four heavy atoms along with their associated hydrogens converge into a single interaction point termed a bead. For peptides, this MARTINI mapping translates the amino acid backbone into a singular bead, specifically the P5 bead, which is its most polar type. This parametrization efficiently captures the behavior of various CP sequences. However, a significant drawback emerges: the omission of interacting groups causes the loss of D/L chirality and the inherent directionality of CP interactions. Consequently, this permits self-assembly in situations where atomistic models would prohibit certain rotations. This leads to an over-representation of self-assembly in MARTINI.
Addressing Directionality: The Introduction of MA(R/S)TINI
MA(R/S)TINI emerges as an innovative adaptation of the MARTINI ForceField, designed to rectify the challenges with directionality while retaining the benefits of Coarse-Grain modeling. Unlike the standard mapping that represents the CP backbone with a single bead, MA(R/S)TINI employs a trio. The primary bead, CPBB, mirrors the properties of the original P5 but has a reduced sigma value for self-interaction, shifting from 0.47 to 0.40. The accompanying beads, CPCO and CPNH, are novel additions exclusively interacting with one another, characterized by σ= 0.26 and ε= 5.6. The spatial arrangement of these beads derives from atomistic model insights. Impressively, this modified parametrization accurately captures both Antiparallel and Parallel interactions. It mirrors permissible rotational angles and trims the CP-CP distance from the initial 5.0 Å to the 4.7 Å, as documented by Ghadiri’s group. Throughout these enhancements, the integrity of CP sequence behaviors remains intact.
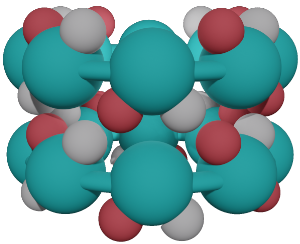
Parallel or Antiparallel?

Antiparallel
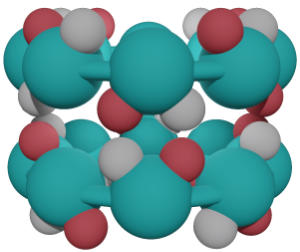
Parallel